 |
|
|
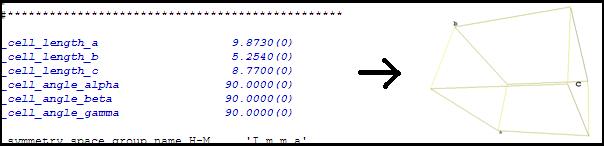
Crystallography tools menu (left-bottom)
Show crystal cell The same
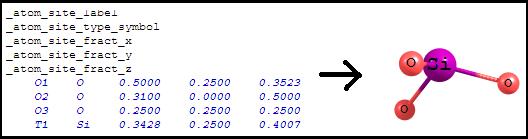
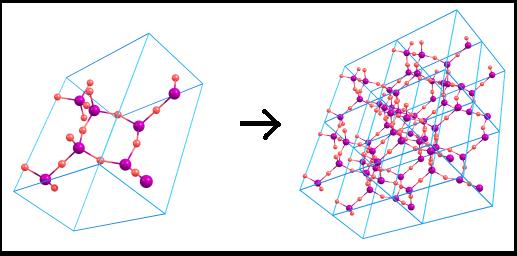
Show
asymmetric
unit
Shows the atoms printed in the .cif file here:
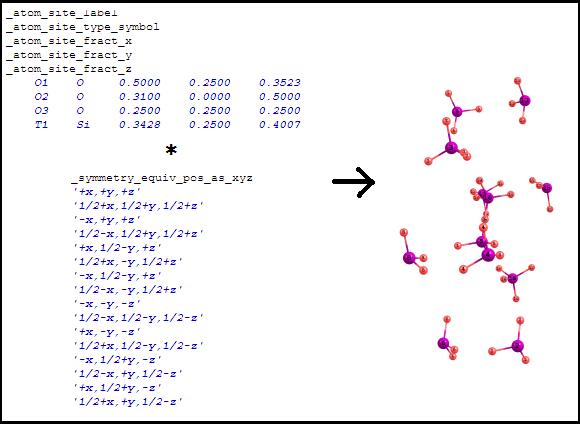
Show
unit cell content (wrapped coordinates) This
is the default display mode. The
coordinates from the asymmetric unit are translated according to all
transformations written in the "_symmetry_equiv_pos_as_xyz"
section. After the coordinates translation, some of the atoms can overlap:
Overlapped atoms are cut by Chemcraft by default. In tools/preferences, you can alter the default distance threshold for cutting. Note
that the
fractional coordinates from the previous point are wrapped, i.e. only
their fractional parts are taken (e.g. 1.15 becomes 0.15, -0.25 becomes
0.75, etc):
Show
symmetry equivalent positions (non-wrapped coordinates)
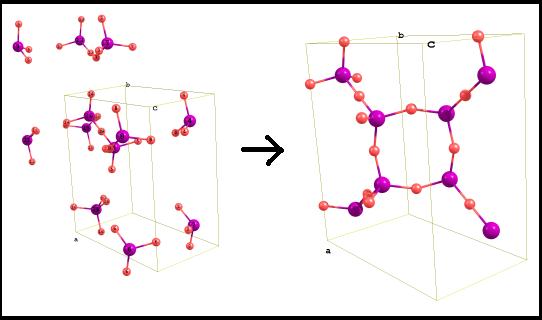
Show
duplicated cell The
crystal cell can be unlimitedly duplicated in different directions:
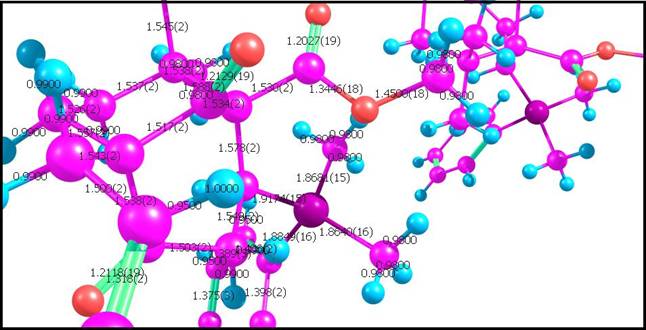
Note that with the subitems of this menu item, you cap specify any size of the duplicated molecule, or the duplicated molecule with non-integer boundaries; Show labels on atoms With this menu item, you can show the numbers of atoms in the default cell for a duplicated molecule; Show structural parameters from the file Some .cif files contain structural parameters (bond lengths, angles, dihedrals, etc). These parameters must be already computed by the programs which generates .cif files. Because in crystallography the position of an atom is not a precise point, but instead a smudge of electron density, spread over a (typically) non-spherical area, these parameters are more accurate, than the parameters, calculated by Chemcraft by the Cartesian coordinates of atoms:
Show structural subunit extracted from 5*5*5 duplicated cell by selected atoms This tool can be a convenient addition to other tools for investigating what atoms are in a specific part of the molecule. It does the same as if you duplicate the cell to 5*5*5 size, then select a file and choose �Tools/Fragments extractor� menu item in the main window; Increase the distance cutoff so there will be no overlapping atoms (closer than 0.3 Angs) If your cif file produces overlapping atoms, choose this menu item. Note that the threshold of 0.3 Angs can be altered via "Tools/Preferences/.cif files/Atom distance threshold for manual cutting" menu of the main window; Wrap fractional coordinates Wraps the coordinates with two possible approaches: Delete atoms with fractional coordinates... This option simply deletes coordinates with e.g. X fractional coordinate lower than 0.0 and bigger than 1.0; Smartly place all atoms into... With this option, an atom with e.g. 1.6 X fractional coordinate will have 0.6 fractional coordinate. And one more check will be done - if two atoms overlap, either first or second will be removed. Also, if there are overlapping atoms with e.g. 0.000001 and 0.999999 X fractional coordinates, the program will remove one of them too with smart check; Convert the atomic coordinates The coordinates of atoms in molecule will be recomputed to fractional ones or vice versa (this is not convenient for visualization, but you can switch to Coord menu and see the values of the fractional coordinate); Calculate bonds by short contacts (VDW radii) Computes the bonds in molecule by Van-Der-Waals radii; Redefine lattice This menu item allows you to rebuild the crystal, describing new A, B, C vectors in terms of old;
Build
slab This menu item builds a
slab (Miller index) useful for investigating a surface of a crystal (e.g.
for heterogeneous catalysis Rebuild cif file by current molecule If you have made any modifications of the molecule's structure on the image in the main windows, you need to use this menu item before saving cif/vasp file (this menu item updates the data stored in the hierarchical list at left-top, which is used for e.g. building duplicated cells); Recompute cell ABC parameters from lattice vectors Some output files contain only the lattice vectors instead of the cell parameter (a, b, etc), and in some programs (Quantum Espresso), the lattice vectors can be presented in an orientation which is considered as non-standard by Chemcraft (e.g. A.y is nonzero). In this case, you need to reorient your molecule before saving a .cif file; Show/edit crystal cell parameters You can use this menu item for altering e.g. the length of the C vector in your crystal; Save .cif or vasp file Saves current crystal data (stored in the hierarchical list at left-top) to a file (also see above about the "Recompute cell ABC parameters" menu item);
Notes: If Chemcraft incorrectly identifies bonds in
molecule, you should use the "Tools/Chemcraft incorrectly
identifies..." menu item or manually set all bonds via Ctrl+B. To make
a bond dotted, double-click on it and alter its type (make it
"H-bond"). If
you work with CIF files, it is recommended to check them via CheckCIF:
| |