 |
|
|
Mass spectrum calculator
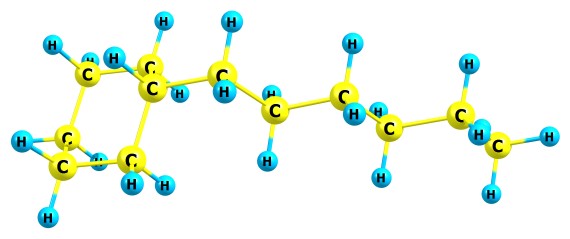
The menu items "Mass spectrum analysis tools" can be useful for chemists dealing with mass spectra: Tools/Mass spectrum analysis tools/Calculate the mass of current molecule with standard distribution of isotopes This utility simply computes the mass of your molecule. You can edit the table with the standard masses of isotopes and their percentages via "Check source data on atomic masses". By default, the file "atomicmasses.txt" in Chemcraft folder is read; this file was generated at NIST Chemistry WebBook site. Tools/Mass spectrum analysis tools/Calculate the mass of current molecule with custom distribution of isotopes With this option, you can specify the isotopes for your molecule. By default, all isotopes are taken for the computation with standard percentages and masses. If you click on an element of the check box list in this window, the program will uncheck other isotopes for this atom. This means that for each atom, either all isotopes are taken with standard percentages, or only one isotope is taken with percentage 100%. Consider you have a CH molecule. The C atom has two isotopes: C12 with m=12.0 and percentage 98.93%, C13 and with m=13.00335 and percentage 1.07%. The H atom has two isotopes: H1 with m=1.00782 and percentage 99.99%, H2 with m=2.0141 and percentage 0.01%. By default (when all checkboxes are checked), the total mass is computed as follows: M=12.0*0.9893+13.00335*0.0107+1.00782*0.9999+2.0141*0.0001=13.0187. If you check the 2H checkbox, the sum will be computed as follows: M=12.0*0.9893+13.00335*0.0107+2.0141*1=14.0248 Checking the checkboxes in this form does not alter the table of standard isotope masses and percentages for the form below. Tools/Mass spectrum analysis tools/Calculate the mass spectrum of fragment ions This utility can compute the peak in mass spectrum of a molecular or fragment ion, with standard isotope masses and percentages (if you want to alter these data, the file "atomicmasses.txt" with them can be edited manually in any text editor). If the checkbox "Set custom molecular formula" is pressed, you can manually set the molecular formula of your compound; however, with this option, the tools like checking rearrangement ions will not be available, since these tools need not only the molecular formula but also the molecular structure (more exactly, the information about the bonds between atoms). With this checkbox, only the menu item "No filter (enumerate all molecular formulas)" can be applied (see below). If the combo box "Compute the mass spectrum of the molecular ion" is chosen, after pressing "Calculate" the program simply computes the mass corresponding to your molecule. After pressing this button, you can click the first line in the list at the center of the window, and then press "Show the mass spectrum of selected fragment ion" and the program will draw a mass spectrum (more exactly, the form of the peak of the molecular ion). The textbox "Broadening" must have the value corresponding to the resolution of your mass spectrometer. If the combo box "Compute the mass spectrum of all fragment ions" is checked, after pressing "Calculate" the program will show a list of all possible fragment ions which correspond to the specified molecular formula. If the combo box "Find possible fragment ions of the specified molecule which can produce the specified peak" is checked, the fragments enumerator will also check whether each found fragment ion produces a peak in the range specified at the "Peak position..." textboxes (otherwise this fragment will not be added to the list). By default, the combo box "No filter (enumerate all molecular formulas)" is set, which means that all combinations of atoms below the molecular formula can be got (for example, if the molecular formula if CH2, the program will show a list of ions CH2, CH, C, H2, H). With this option, the calculation is performed relatively fast: for example, for C32N8H18 molecule, the number of steps will be 33*9*19=5643 (from 0 to 32 carbon atoms, 0-8 N atoms and 0-18 H atoms). However in some cases this enumeration is not sufficient. For example, you have a mass spectrum of this molecule C12H24:
This molecule can (theoretically) produce two different fragment ions with same molecular formula C6H12 and mass 84.16:
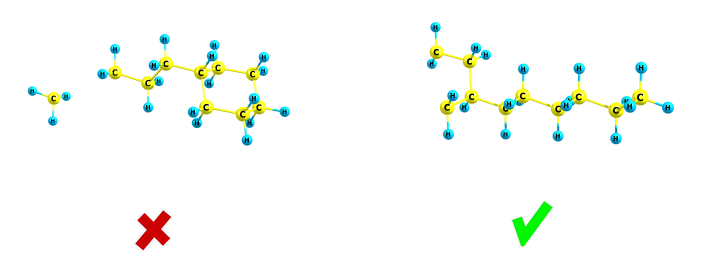
In some cases you will need to check all such fragment ions, and for this, you can use the second menu item "No filter (enumerate all combinations of atoms, 2^N time)". This option simply enumerates all combinations of atoms in your molecule (each atom can be either present or not present in each fragment). And if you choose this menu item, the option "Add different ions with same molecular formula" can be checked, so for the example above both fragments will be added to the list below. And with this option, you will be also able to see the molecular structure of each fragment ion, with the button "Show the image of the selected fragment ion" (which becomes active after clicking the list box with fragment ions at the center of the window). The next button "Show for all" shows all ions present in the list (as a .xyz file with multiple structures). This option "No filter (enumerate all combinations of atoms, 2^N time)" works too slow for big molecules (because the number of all combinations 2^N, where N is the number of atoms, is too big for N>25). If the combo box "Same but exclude all rearrangement ions" is checked, the program will similarly enumerate all atomic combinations, but the fragments which become isolated will be discarded:
If the combo box "Recombine ions after bond removal" is checked, the algorithn of ions enumeration will work as follows. The program will remove arbitrary bonds in your molecule (by default - all possible triples of bonds which are single), and check what fragments appear, and then recombine these fragments. Consider you have this molecule: When the program removes the bonds C1-C11 and C17-C21 during the work of the algorithm, the following recombination ions will be possible:
The last 3 fragments will be added to the list only if the option "Customize bonds removal options/Recombine fragments" is enabled. Note that if your molecule contains aromatic cycles, and you want less aromatic bonds to be removed by the algorithm, you should set the auto-determination of aromatic bonds via "Tools/Chemcraft incorrectly identifies bonds in your molecule?/Identify bond types by atom valence information (aromatic bonds)". Possibly you should also switch on the "Display/Quick" display scheme since this scheme shows whether a bond is single or aromatic or double:
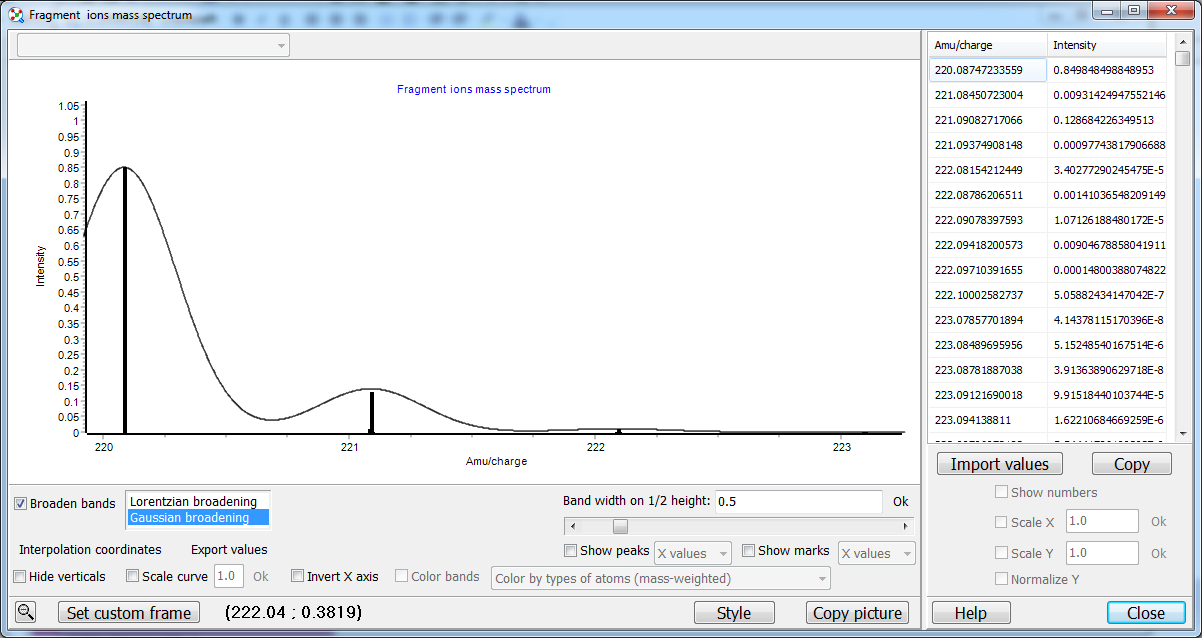
You can also set the bonds in your molecule manually for your purposes. These options can be useful since Chemcraft allows to specify the maximum number of single bonds which will be removed by the fragments enumerator, and the maximum number of non-single bonds (aromatic, double, triple...). For aromatic cycles, possibly often it is useful to specify the limit of non-single bonds to be removed to 1, which means that the aromatic cycles can't be broken during the ionization in the mass spectrometer. If your mass spectrometer has a high resolution and shows individual isotopes of each fragment ion, you should click "Show the mass spectrum of the selected fragment ion" to see the shape of the fragent ion peak:
Sorry if you find this ad strange, here is a screenshot of a message from Chemcraft user to our support:
| |