 |
|
|
Working with crystallography filesThe
following information can be extracted from crystallography information
files (.cif): 1)
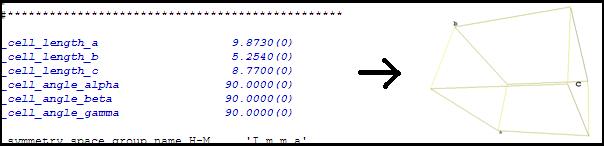
Cell parameters (a, b, c, alpha, beta, gamma).
The
menu items "Show lattice vectors" and "Show crystal
cell" are used to visualize cell parameters. 2)
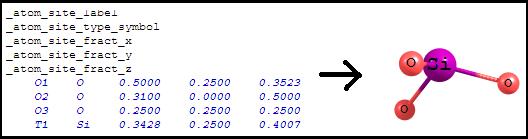
Asymmetric unit.
3)
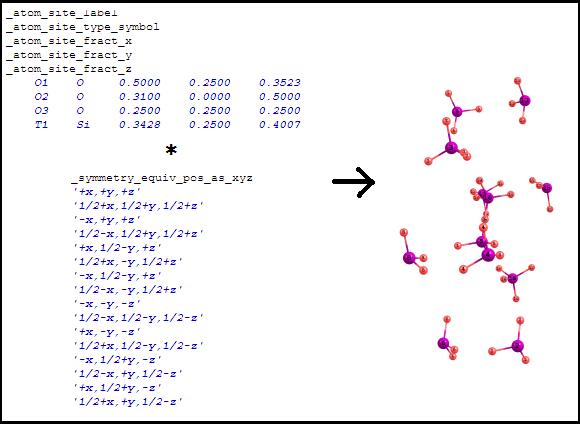
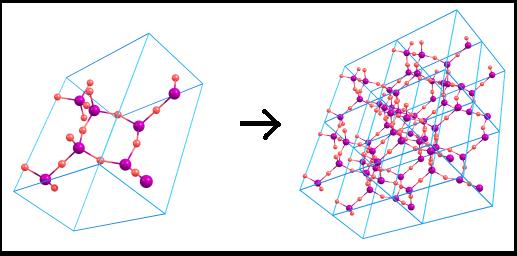
Symmetry equivalent positions (non-normalized). The
coordinates from the asymmetric unit are translated according to all
transformations written in the "_symmetry_equiv_pos_as_xyz"
section. After the coordinates translation, some of the atoms can overlap:
4)
Coordinates normalization. The
fractional coordinates from the previous point are normalized, i.e. only
their fractional parts are taken (e.g. 1.15 becomes 0.15, -0.25 becomes
0.75, etc):
5)
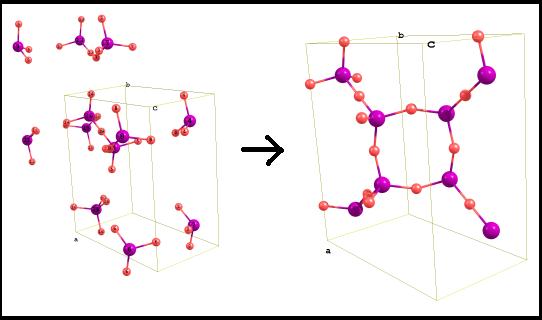
Coordinates duplication The
crystal cell can be unlimitedly duplicated in different directions:
6)
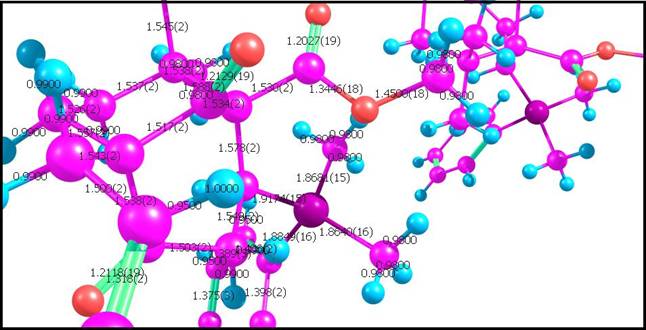
Structural parameters from the file: Some
.cif files contain structural parameters (bond lengths, angles, dihedrals,
etc). These parameters must be already computed by the programs which
generates .cif files. Because in crystallography the position of an atom is
not a precise point, but instead a smudge of electron density, spread over a
(typically) non-spherical area, these parameters are more accurate, than the
parameters, calculated by Chemcraft by the Cartesian coordinates of atoms.
Note: if Chemcraft incorrectly identifies bonds in
molecule, you should use the "Tools/Chemcraft incorrectly
identifies..." menu item or manually set all bonds via Ctrl+B. To make
a bond dotted, double-click on it and alter its type (make it
"H-bond"). If
you work with CIF files, we recommend checking them via CheckCIF:
| |