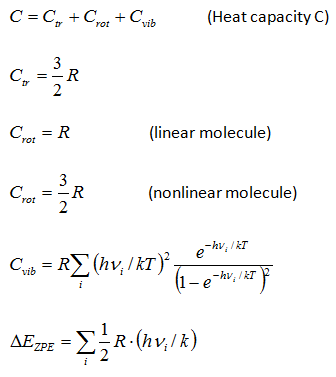 |
|
|
Thermodynamic functions calculatorChemcraft can compute the entropy and the Gibbs energy of a molecule by its molecular parameters, including the geometry (moments of inertia) and vibrational frequencies. This computation is made in "rigid rotor - harmonic oscillator" approximation (RRHO). The main aim of this utility is the possibility to cut low frequencies, which produce big errors because of ahharmonicity. You can specify a threshold value, e.g. 100 cm**-1, and the values of the frequencies below this threshold will be increased to this threshold value. The formulas for the computation were taken from the following refs.:
ASE thermochemistry computation. https://wiki.fysik.dtu.dk/ase/ase/thermochemistry/thermochemistry.html
You can download the Delphi source code which computes the thermodynamic functions in Chemcraft here: chemdata_u_b768.zip. Troubleshooting The rotational entropy is computed by Chemcraft using the masses of atoms corresponding to most abundant isotopes (e.g. 12.0 for C, 1.007825 for H, etc). Some disagreement can occur if in your input file non-standard isotopes were specified (e.g. 13.003355 for C). In this case, you should obtain a Hessian (from Gaussian .fch file or Orca .hess file) and compute the frequencies setting the non-standard isotopes, and then compute the Gibbs energy using this tool (don't forget to manually uncheck the 6 frequencies corresponding to rotational and translational motion). Versions of Chemcraft from b648 to b763 didn't compute the electronic entropy Sel=ln(M); because of that, the Gibbs energy computed with them didn't match the Gibbs energy computed by Gaussian or Orca for molecules with multiplicity other than 1 (if M=1, the electronic entropy is zero).
Experimental verification of the Gibbs energies computation
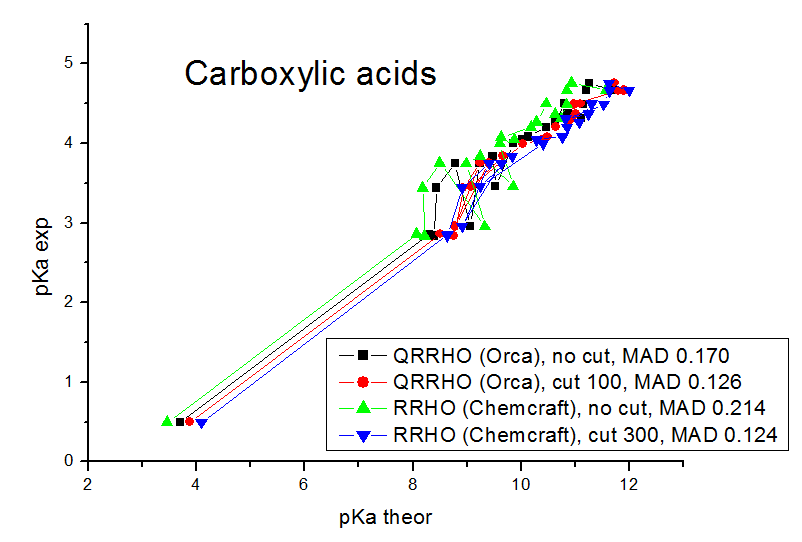
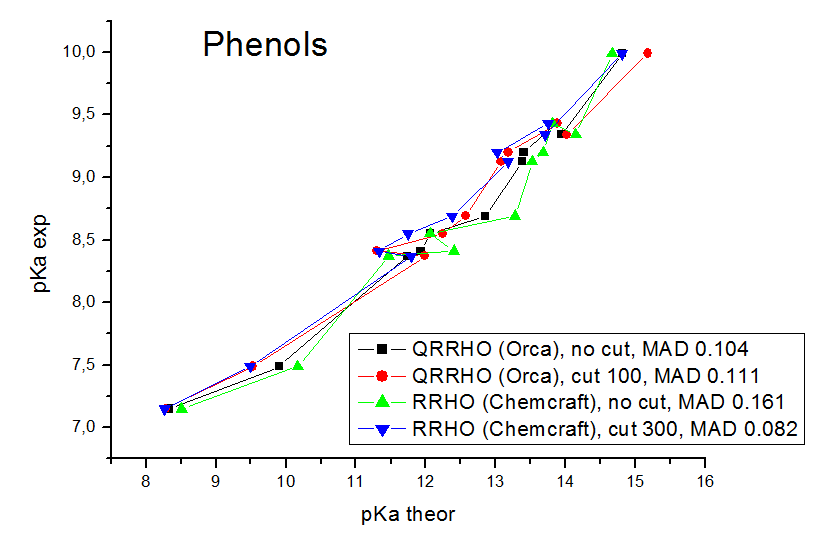
Our models contain explicit water molecules which produce very small frequencies (like 10 cm**-1), and we found that the anharmonic origin of these frequencies produces very big errors. The mean absolute deviations (mean differences between the experimental pKa values and the values calculated by linear fit parameters) are in the range of 0.08 - 0.17 pKa units. For the computations of Gibbs energies and pKa values, the MADs for the carboxylic acids obtained are 0.170 pKa units for Orca QRRHO computations without the low frequency threshold, 0.126 pKa units for Orca QRRHO computation with 100 cm**-1 threshold, 0.214 pKa units for Chemcraft RRHO computation without the threshold, 0.149 for Chemcraft RRHO computation with the threshold 40 cm**-1, 0.144 with 100 cm**-1 threshold, 0.124 with 300 cm**-1 threshold, and 0.126 with 800 cm**-1 threshold. For RRHO computations of phenols, the MADs are 0.104 pKa units for Orca QRRHO computations without the low frequency threshold, 0.111 pKa units for Orca QRRHO computation with 100 cm**-1 threshold, 0.161 pKa units for Chemcraft RRHO computation without the threshold, 0.118 for Chemcraft RRHO computation with the threshold 40 cm**-1, 0.090 with 100 cm**-1 threshold, 0.082 with 300 cm**-1 threshold, and 0.081 with 800 cm**-1 threshold. So, our study confirms that applying the threshold of 100-800 cm**-1 is a good approach which improves the agreement of computed Gibbs energies with the experiment. At these pictures you can see the comparison of correlations between the computed and experimental pKa values obtained in our study with different approaches:
We have published our results in Molecular Physics (DOI 10.1080/00268976.2024.2396535): https://simplecompchem.blogspot.com/2024/09/qspr-prediction-of-acidities-of.html Some people can find somewhat strange, that the best agreement with experiment in our study was achieved with 300 cm**-1 and 800 cm**-1 thresholds, while it is usually recommended to use the value of 100 cm**-1 threshold. Possibly this is a statistical deviation caused by the fact that the number of compounds is not big enough, and, more importantly, there are much more significant sources of errors here (the main source of errors in an incomplete taking into account of the specific solvatation). We found that with the 800 cm-1 thresholds, the agreement with the experiment (MADS for linear fits) is almost the same as if no entropy computation is performed (when the Kohn-Sham energies of deprotonation are correlated with the pKa values, not the computed pKa values). This means that the role of entropy computations is almost neglible, and still if these computations are performed without the thresholds - an additional error appears due to the neglect of the anharmonicity of small modes.
Citation We suppose that a proper citation can be like this: "The Gibbs energies were computed with Chemcraft program [1], in which the commonly used formulas for RRHO approach [2] have been implemented. An approach of raising low vibrational frequencies to a specified threshold value, firstly suggested by Truhlar et all [3], was used." [1] See the Citation page [2] Donald A. McQuarrie, John D. Simon. Molecular Thermodynamics. University Science Books, 1999. [3]
Ribeiro, R. F.; Marenich,
A. V.; Cramer, C. J. & Truhlar, D. G. "Use of Solution-Phase
Vibrational Frequencies in Continuum Models for the Free Energy of Solvation”.
J. Phys. Chem. B 2011, 115, 49, 14556–14562
| |