|
| | ||||||
| Main | News | Description | Download | Contacts | Help | Blog | |
| Order | Update | Gallery | About Us | Links | Forum | Citation | |
|
Some
tutorials on quantum chemistry and scripting “magic” in Chemcraft
Many people say that the quantum chemistry has a low prediction power. Our 20-years experience of performing research work using QC computations confirms this, but this statement must be explained as follows: usually, when you directly compute some property with QC methods, the error of the result is very big, but if you have a set of homologues and compute some properties of them, usually these compute properties correlate well with its properties measured experimentally. Below are some examples from our research work.
Systematic
errors in quantum chemistry 1.
Let’s consider you want to know the pK of an acid, for example the benzoic
acid:
If you have very old software for performing QC computations like Gaussian94, and know almost nothing about quantum chemistry, you can still obtain some results: for example, you can compute the energy of deprotonation of this molecule in gas phase. For obtaining this energy, you need to compute the energy of this molecule and also the energy of its deprononated anion:
Since we consider that the proton has zero energy, the energy of
deprotonation is the difference between the total SCF energies of deprotonated
anion and the neutral molecule. With B3LYP/6-31++G(D,P) method, this energy is
0.5517 a.u. or 1448.6 kJ/Mol. The physical meaning of this energy is that
one needs to spend 1448.6 kJ/Mol for tearing off one proton from a molecule
of benzoic acid in gas phase. To compute this energy, you need to run 2 e.g. Gaussian jobs with the following input files:
#P
B3LYP/6-31++G(D,P) OPT Test
for tutorial 0
1 C
0.222444000000
0.036011000000
-0.000062000000 C
-0.522231000000
1.238748000000
-0.000047000000 C
-0.444080000000
-1.211392000000
-0.000034000000 C
-1.926266000000
1.193822000000
-0.000016000000 C
-1.849364000000
-1.251243000000
-0.000001000000 C
-2.591409000000
-0.051172000000
0.000008000000 H
0.004303000000 2.188608000000
-0.000065000000 H
0.133765000000
-2.129254000000
-0.000045000000 H
-2.499100000000
2.116828000000
-0.000009000000 H
-2.363687000000
-2.208115000000
0.000016000000 H
-3.677775000000
-0.085331000000
0.000033000000 C
1.703734000000
0.126730000000
-0.000106000000 O
2.363066000000
1.189155000000
0.000102000000 O
2.328992000000
-1.108381000000
0.000075000000 H
3.309071000000
-1.017949000000
0.000202000000
#P
B3LYP/6-31++G(D,P) OPT Test
for tutorial -1
1 C
-0.278240000000
0.000004000000
0.000000000000 C
0.439059000000
1.217813000000
0.000498000000 C
0.439060000000
-1.217811000000
-0.000496000000 C
1.847823000000
1.220201000000
0.000563000000 C
1.847822000000
-1.220203000000
-0.000564000000 C
2.556945000000
0.000000000000
-0.000001000000 H
-0.120813000000
2.148282000000
0.000827000000 H
-0.120815000000
-2.148279000000
-0.000823000000 H
2.390726000000
2.162847000000
0.001043000000 H
2.390728000000
-2.162847000000
-0.001044000000 H
3.644616000000
0.000000000000
-0.000001000000 C
-1.809369000000
-0.000013000000
0.000002000000 O
-2.402689000000
1.152920000000
-0.001279000000 O
-2.402690000000
-1.152913000000
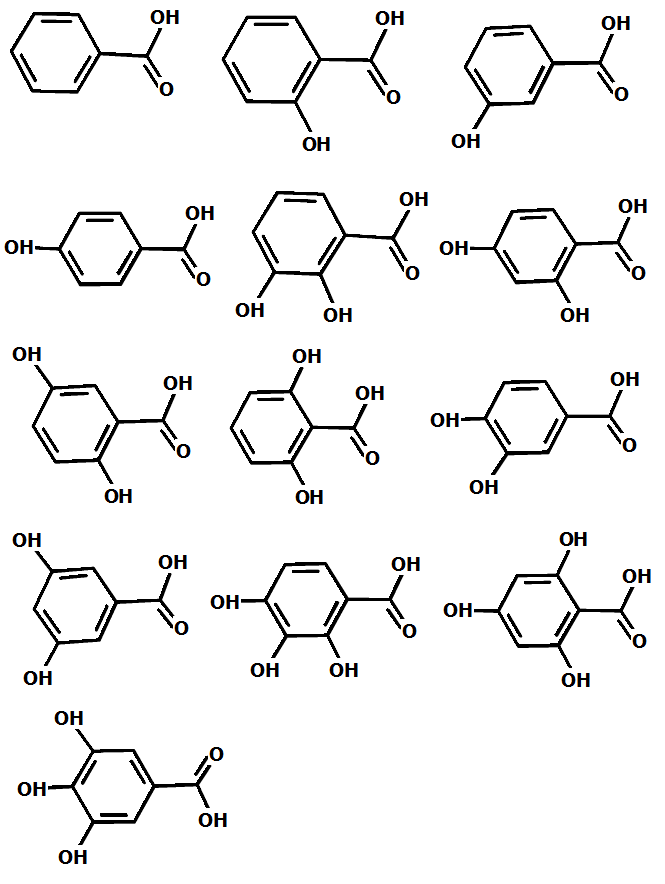
0.001277000000 The value of the energy of deprotonation itself gives you almost no information about the required property – pKa of the benzoic acid in water. But let’s consider you have 13 homologues of the benzoic acid:
It is intuitively clear that the energy of deprotonation of these compounds
must correlate with the pK: the lower this energy, the easier it is for the
acid to release a proton, and accordingly, the higher its acidity (or lower
the pKa value). If these energies are computed for all 13 molecules (it is
important to use the same method and the same level of accuracy for all
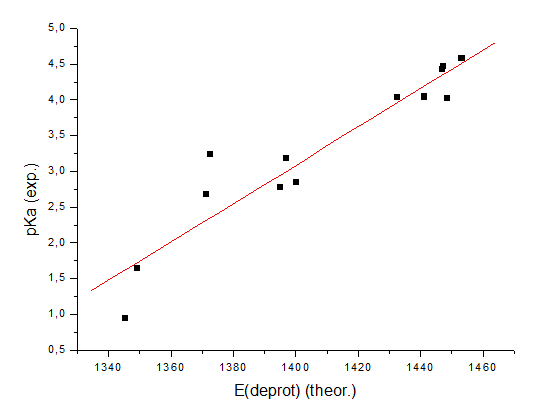
computations), the following correlation of the computed energies and
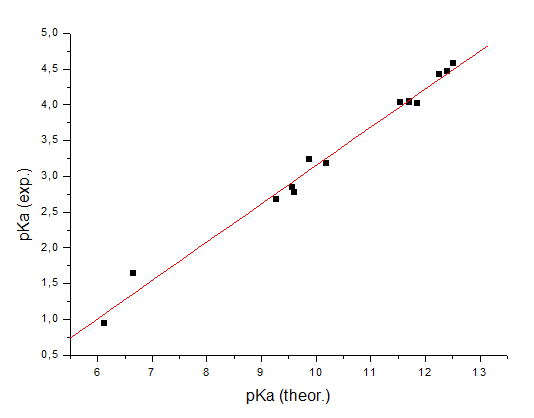
experimental pKa values in water can be built:
To
improve this correlation in our work (DOI:10.1080/00268976.2024.2396535 -
Slightly better level of computation (B3LYP-D3/aug-cc-pVTZ); -
Modeling water solvent via implicit-explicit approach (PCM model and
explicit water molecules); -
Direct computation of Gibbs energies instead of SCF energies (in other
words, taking into account the entropic factor); -
Raising low frequencies to avoid errors caused by anharmonic nature of low
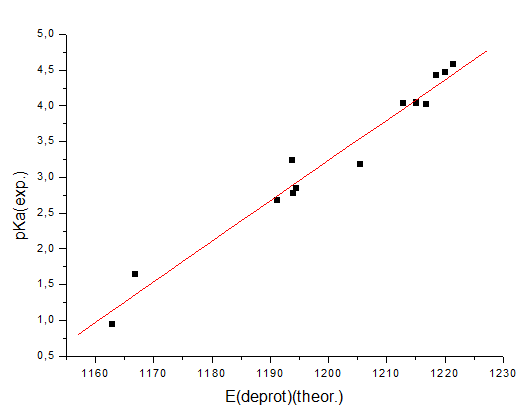
vibrational modes. With these improvements, we have obtained the following correlation:
As you can see, the correlation has become significantly better. Since we
had computed the entropies and Gibbs energies of these compounds, the X
values in the graph are the pKa values instead of energies; but as you can
see, their absolute values are still too far from the experimental ones (the
systematic error is great). This is a common situation in quantum chemistry.
At this
post in our blog you can see many other similar correlations between the
computed and experimental properties, and usually the systematic error is
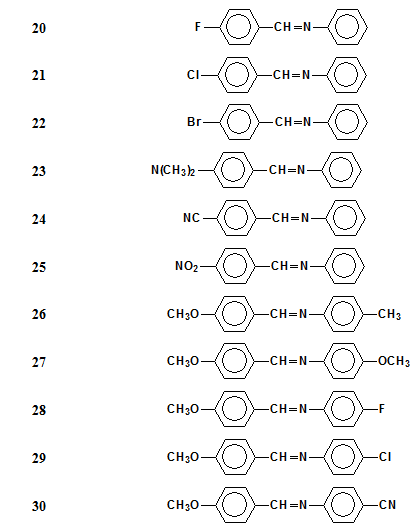
really big. 2. Here is one more example from our research work, which illustrates the low predicting power of quantum chemistry. We studied the properties of a set of 45 liquid crystals (nematic Schiff bases) like these ones:
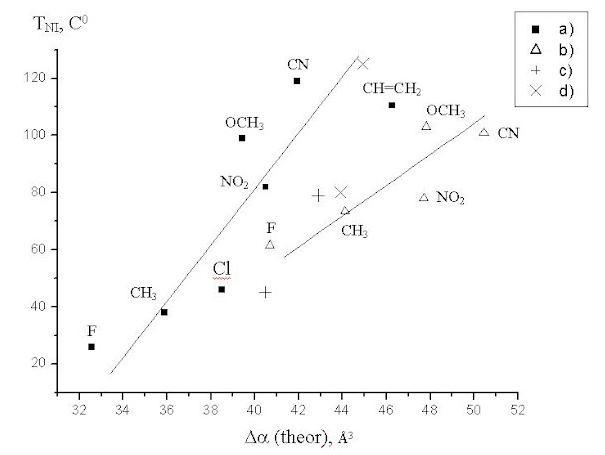
The property which has practical use is the thermal stability of these liquid crystals, or the temperature of phase transition between the nematic and isotropic state. Our knowledge of computational chemistry, our experience and intuition clearly indicate that it is almost impossible to directly predict this value via quantum chemistry methods: those who somehow predict such properties get PHDs in technical sciences, not in chemistry. But there is a theory that the thermal stability must correlate with the anisotropy of molecular polarizability of these molecules. To check this, we computed the polarizabilities of these molecules via quantum chemistry (B3LYP/6-31G(D,P)) and drew the correlation between them and the experimental temperatures of phase transition:
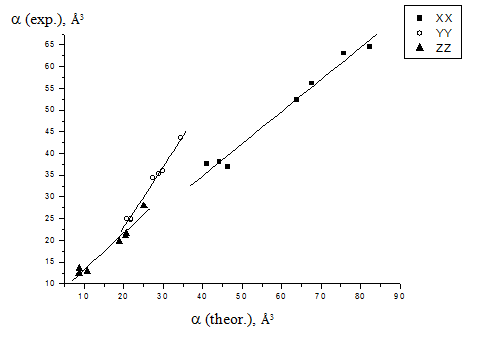
As you can see, the correlation is very bad, but it exists. To exclude the presumption that this correlation is bad because the accuracy of our quantum chemistry computations is very low, we also drew the correlation between the computed and experimental (from literature) values of the components of the polatizability tensor of our compounds:
You can see here that the correlation is good, but still there is rather big
systematic error of the computations, as one more example.
Using correlations in your research work
The examples above illustrate the following: if you want to compute some
property of your system via quantum chemistry methods, usually it is not a
good idea to compute it directly. But you can choose a set of similar
compounds and compute the properties of them, and draw the correlation
between these computed properties and experimental ones. We think that the
“art” of quantum chemistry is to take several compounds with well-known
experimental properties, check the correlation of them with the computed
properties, and then add to this correlation a compound which is mostly
important for your research, and check whether this compound is an outliner
in the correlation for making important conclusions. We used exactly this
approach when studying the chemisorption of carbon monoxide on nickel oxide
(DOI:10.1016/j.molstruc.2021.130439
If you want to explain these ideas and this approach in your publication,
you can cite e.g. the following phrase from the book "pKa Prediction
for Organic Acids and Bases
The
most useful methods of pK. prediction are based on the assumption that
within particular classes of acids and bases, substituents produce free
energy changes which are linearly additive. This assumption has led to many
published Hammett and Taft equations which are given in the Appendix
together with extensive tables of substituent constants. A large number of
worked examples using these equations, and other predictive methods occur
throughout the text.
Why
Chemcraft significantly facilitates computing a set of compounds The aim of Chemcraft is to reduce the time needed for performing the QC computations in your study; and besides our old features like molecular constructor, symmetrizer etc., the latest versions of Chemcraft comprise a set of scripting tools for automated working with many files at once. Here is how you can use them.
Extracting
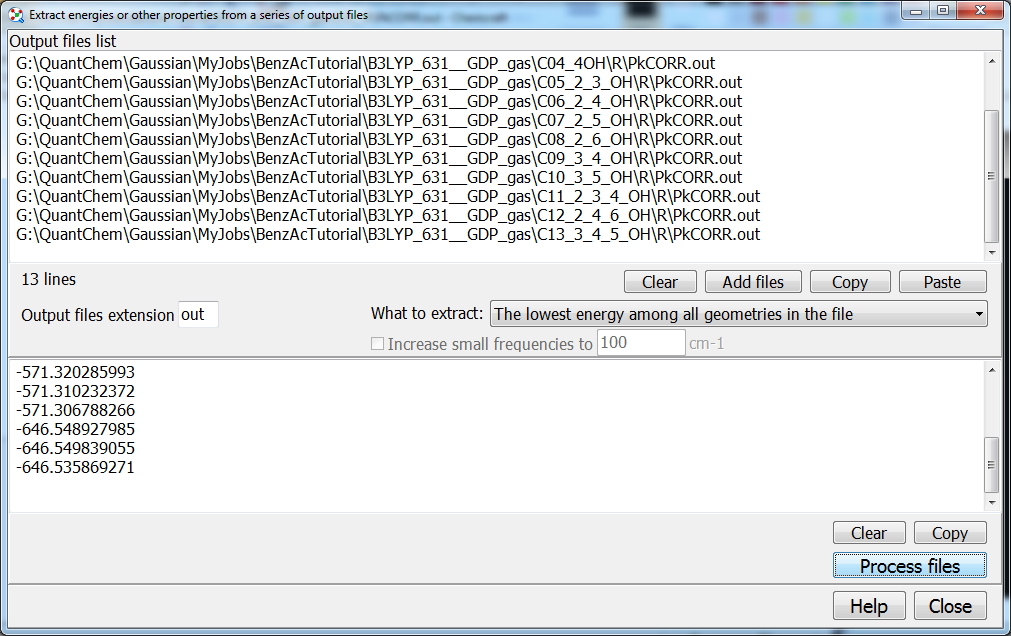
energies or other properties from multiple output files Let’s consider you have computed 13 benzoic acids above: each compound requires two jobs of geometry optimization - neutral molecule and anion. To most quickly obtain 13 energies of deprotonation (the differences between the energies of molecule and anion), you can paste two columns into Excel or Origin. For getting these two columns, you can use the “Tools/Scripts/Extract energies from multiple output files” utility in Chemcraft:
In this window, you need to enter the file names of all output files with computed energies, or maybe Gibbs energies, or other properties. The file names are entered in text format, so you can use any tool for getting or editing this text; with Chemcraft, you can sequentially click many files in Windows Explorer for getting this list, or you can select a folder and all files inside this folder will be added to this list.
Modifying
multiple input files
Let’s consider you have computed the energies of a 13 homologues of
benzoic acids enlisted above, at B3LYP/6-31++G(D,P) level. Now you want to
recalculate these molecules with a better level of theory – switching from
B3LYP to B3LYP-D3 and adding implicit solvation model (PCM). The input files
will look as follows: #P
B3LYP/6-31++G(D,P) OPT scrf(pcm,solvent=water) EmpiricalDispersion=GD3 Test
for tutorial 0
1 C
0.220241000000
0.030476000000
0.000007000000 C
-0.514968000000
1.225419000000
-0.000017000000 C
-0.448283000000
-1.203510000000
0.000008000000 C
-1.908125000000
1.186335000000
-0.000040000000 C
-1.843137000000
-1.236882000000
-0.000015000000 C
-2.573446000000
-0.044458000000
-0.000039000000 H
0.021009000000
2.168560000000
-0.000017000000 H
0.123933000000
-2.124111000000
0.000026000000 H
-2.475255000000
2.112287000000
-0.000059000000 H
-2.360036000000
-2.191837000000
-0.000014000000 H
-3.659299000000
-0.074001000000
-0.000057000000 C
1.704089000000
0.122335000000
0.000034000000 O
2.339996000000
1.159905000000
0.000027000000 O
2.317277000000
-1.091313000000
0.000031000000 H
3.273232000000
-0.917930000000
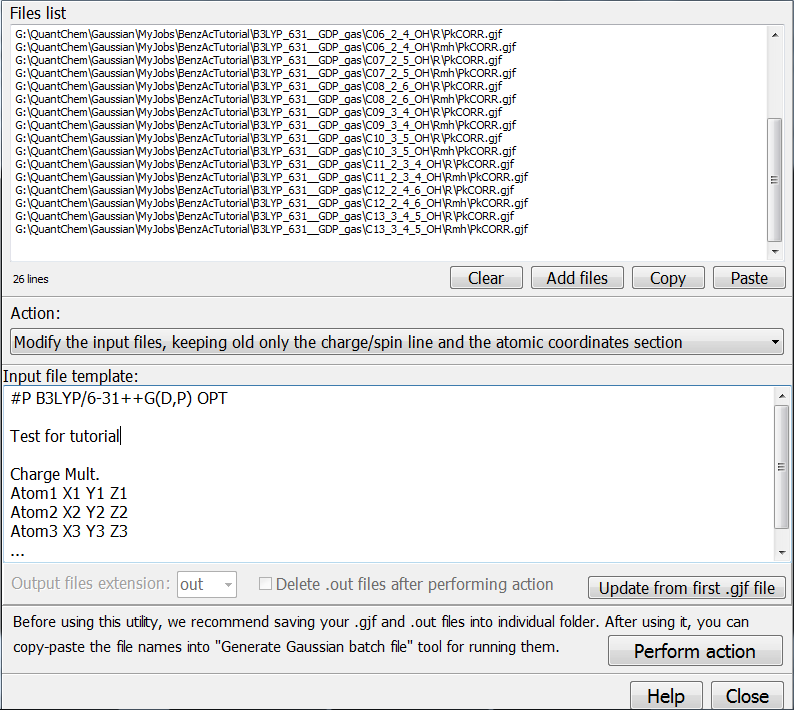
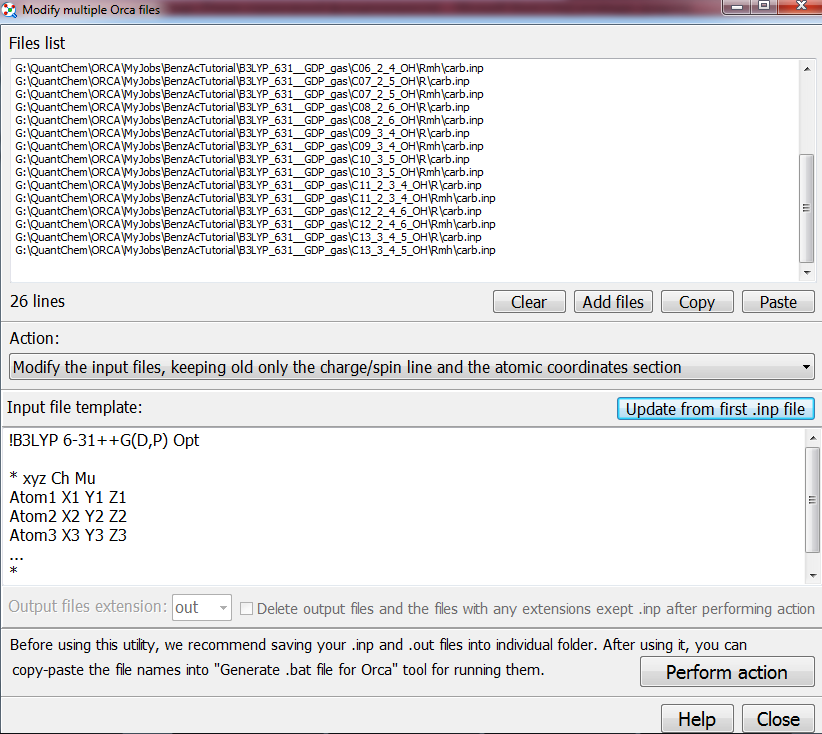
0.000034000000 Multiple input files can be modified with “Tools/Scripts/Modify multiple … input files”:
To redo the computation with a new level of theory, firstly copy all input
and output files with previous calculations into separate folder. After
that, it is often a good idea to firstly replace the coordinates in the
input files with the coordinates read from corresponding output files, from
final geometries with lowest energies. This is done with the choice
“Extract the minimum energy geometries..” from the “Action” textbox
in this window. For each input file, Chemcraft will find the corresponding
output file (with same name but .out extension), extract the coordinate from
the output file, and insert them into input file. Usually the output files
are deleted after this action (see corresponding checkbox).
Then
you need to modify these input files. Switch to “Modify input files…”
option, and then press the button “Update from first .gjf file”. In the
“Input file template” textbox, the first file from the list above will
be shown. Then you can make any modifications in this file: usually only the
route section should be changed, but you can paste there any input file,
including an input file with multiple jobs.
Then,
after pressing “Perform action” button,
all input files from the list above will be modified: Chemcraft will replace
them with the contents of the “Input file template” textbox, except the
section with charge/multiplicity and the atomic coordinates – this section
will not be modified. Chemcraft supports modifying multiple multy-job input
files, at least the ones where the first job contains this section with
charge/multiplicity and coordinates. Versions of Chemcraft earlier than b804
do not show this section in the “Input file template” textbox, but they
work the same as newer versions. With newer versions, in the “Input file
template” textbox, both usual contents of the file can be present for
further applying, or standard lines “Charge Mult. Atom1 etc.”.
After
updating the computation method for compounds above, the correlation becomes
as follows:
As
you can see, the agreement with the experiment has become better, mainly because of the PCM solvation model.
Running
multiple input files
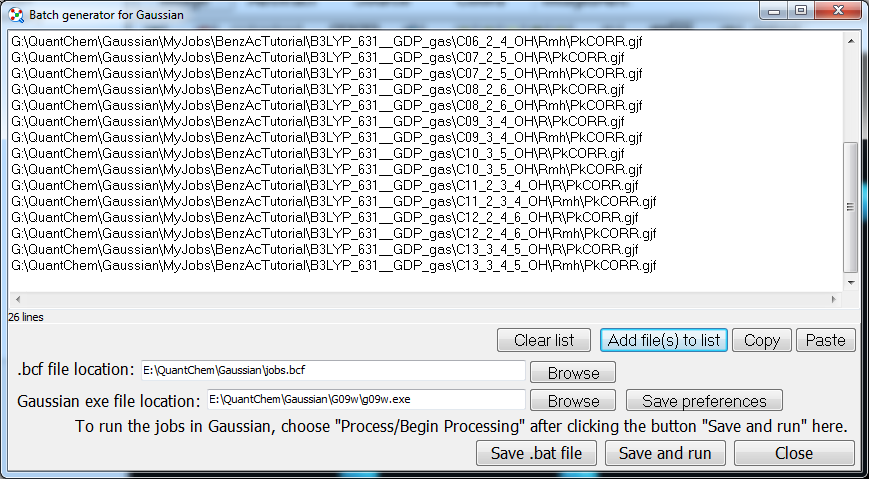
The
utility “Tools/Generate Gaussian batch file” generates a
.bcf file for running multiple input files in Gaussian. Since Chemcraft
works with the lists of file names in text format, you can copy the list of
files from the window above into this window. Besides Gaussian, Chemcraft
creates .bat files for Orca an Gamess, which run Orca or Gamess for each
input file. Custom scripting tools are also available (they can be used for
Linux or Mac) – “Tools/Scripts/Generate custom script…”:
Scripting
tools for Orca The tools above are presented for the case of using Gaussian, but for Orca it is almost the same; the only difference is that for Orca, when you take coordinates from output files and insert them into input files, you can make Chemcraft delete not only the files with .out extensions, but with all other extensions (.hess, .gbw, .xyz, etc.):
Computing
the relative energies and percentages of different conformers
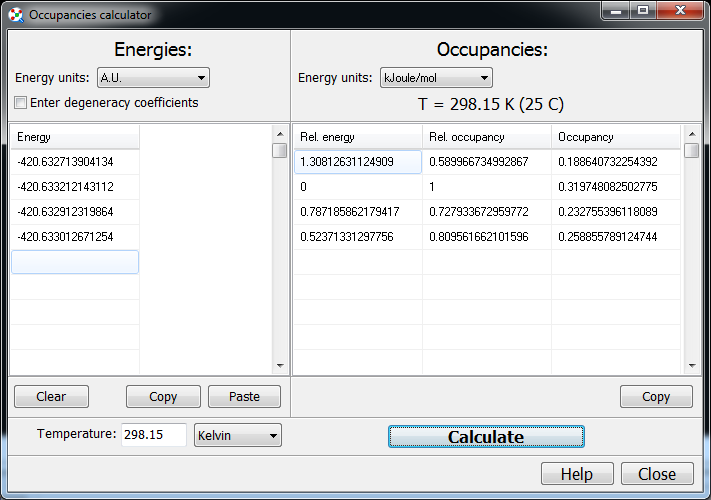
If you have a set of conformers of your molecule, you can compute their
relative energies, and percentages (according to Boltzmann), with
“Tools/Simple utilities/Calculate occupancies of percentages…”:
Again, if you have computed several conformers, you can quickly make
Chemcraft open them and print a column with their energies, and then copy
this column into this window.
| |||||||